|
ICH
ICH Q12《医药产品生命周期管理的技术和法规考量》于6月份结束第三阶段。计划于2019年下半年进入到第四步批准阶段。
这一新指南的提出为构建产品全生命周期管理框架提供指导,促进整个产品生命周期内以更可预测和更有效的方式管理批准后的CMC(化学、制造、控制)变更。 该指南的采用将促进创新和持续改进,并加强产品的质量保证和可靠供应,包括前瞻性地规划供应链调整。 同时也让监管人员(审评员和检查员)更好地了解企业的药品质量体系对批准后的CMC变更的管理。 该新指南旨在补充现有的ICH Q8至Q11指南,包括核心指南和附件,ICH Q12着重在产品生命周期中的商业化阶段。
ICH Q12 草案内容详见:
详情链接
FDA
2019.2 FDA发布《连续制造(Continuous Manufacturing)的质量考虑》指南草案,强调医药产品连续制造中的质量因素。 涵盖了与开发、工艺验证、上市授权和日常制造相关的主题。FDA将连续制造定义为由至少两个连接的操作单元构成、物料连续进料并转化、经过处理的物料(产品)被连续地从系统中移除的过程。传统的批次制造由一系列单独的工艺步骤组成。对于制药行业中新型的集成的、连续制造程序,该指南描述了在连续制造中必须考虑的关键要素。这些要素在各个章节予以介绍:
A.连续制造中的关键概念
-
过程动力学
-
为连续制造过程定义批次
B.控制策略
-
输入物料控制
-
过程监测与控制
-
物料导流
-
实时放行测试
-
质量标准
-
设备
-
系统集成、数据处理和管理
C.工艺验证
-
第一阶段:工艺设计
-
第二阶段:工艺确认
-
第三阶段:持续工艺确认
D.其他药品质量体系考虑因素
E.放大
F.稳定性
G.将现有批次过渡到连续制造
该指南草案适用的重点是固体医药产品或小分子医药产品的制造商。 其原则也可以用于生物制剂的制造。
指南草案内容详见:详情链接
在美国,医疗器械由FDA 器械及放射卫生中心(CDHR)监管。如何快速熟悉医疗器械这个话题并及时获取前沿动态?FDA为此提供了一个单独的网站: CDRH Learn。该网站提供非常全面的信息,包含医疗器械监管框架(幻灯片最近于2019年4月修订)的基础知识、费用要求、UDI、注册问题(场地及产品)、上市许可问题以及商业阶段(上市后活动)的监控要求等。在“上市后活动”标题下,还可以找到关于质量管理体系典型要素的信息,如设计控制、工艺验证、纠正与预防措施体系和投诉等内容。
详细信息可参阅FDA CDRH Learn 网站:详情链接
2019年7月31日, FDA 颁布了《FDA检查医疗器械企业后的非约束性反馈》指南草案。在指南草案中,FDA通知他们将来在对医疗器械制造商的检查后会提供(非约束性)反馈。被检查企业可向FDA发出反馈申请,经审核申请后,FDA将在45个工作日内回复。FDA的非约束性反馈应阐述针对检查缺提出的措施是否充分、部分充分或不充分。非约束性反馈旨在帮助被检查公司处理检查缺陷,但正如指南的名称所示,不具有约束力。
指南草案内容详见:详情链接
美国 FDA 于 7 月 10 日发布《辅料数据库使用指南》草案,解释了即将对 FDA 辅料数据库(IID)开展的一些改进,包括将增加每种辅料的最大每日暴露量(maximum daily exposure,MDE)以及对数据库更频繁地更新。指南还介绍了如何使用 IID 及其局限性。指南表示: “将根据 GDUFA II 承诺函在未来对 IID 进行改进。到 2020 年 10 月 1 日,FDA 将完成对辅料数据库的改善提升,以便使用者可以进行电子查询,获得每种给药途径的最大每日摄入量(maximum daily intake,MDI)和最大每日暴露量信息。” 多年来申请者最关心的问题与最大含量和 MDE 之间的差异相关。最大含量是一个剂量单位中的辅料量,而 MDE 则基于药品标签中建议的给药剂量计算出来。申请者没有确定 MDE 的好方法,是因为 IID 中没有与实际产品的链接。FDA 在 GDUFA II 下的承诺之一是在 2020 年 10 月 1 日之前更新 IID 以包括 MDE。这将是 IID 自建立以来的最大改进。FDA 辅料数据库包括辅料的以下信息:成分名称、给药途径、剂型、CAS号、独特成分标识符和最大含量。工业界可能会使用 IID 中的信息来支持辅料的安全性。被列入 IID 中意味着该辅料先前已用于 FDA 批准的药品中。如果一个辅料已在获批药品中用于特定给药途径,则该辅料通常不被认为是新辅料,并且可以在下次以相同的给药途径用于新产品中时,进行较少的评估。
指南草案内容详见:详情链接
FDA 持续更新警告信,其中以下方面受到关注
-
质量部门的职责
Rxhomeo Private Limited 警告信:
公司的质量部门未能履行职责,以确保生产的药品符合cGMP,并符合既定的特性、效力、质量和纯度的标准。质量部门未能对生产过程提供足够的监督。 质量保证QA-004-00规程没有定义和描述质量部门的结构、功能和职责。警告信内容详见: 详情链接
西安住邦无纺布制品有限公司警告信:
公司未能建立适当的质量部门,该部门的职责和权力是批准或拒绝所有组件、药品容器、密闭构件、中间物料、包装材料、标签和产品(21 CFR 211.22(a))。公司缺乏适当的质量部门。 例如,质量部门未能为多个职责建立书面规程,例如CGMP培训、年度产品回顾(APR)和稳定性计划。警告信内容详见: 详情链接
-
清洁验证:
Laboratoires Clarins警告信
FDA要求:
公司无法证明使用清洁/消毒程序可以去除污染物。 应用的唯一措施是对表面的目视检查。
-
评估清洁/消毒程序和活动的详细计划(清洁/消毒方法,试剂,作用时间,检测方法和可接受标准)
-
清洁/消毒验证策略的科学依据
-
清洁/消毒验证方案更新的总结,包括最坏情况:
-
评估具有最高毒性的产品
-
考虑药物在清洁剂中的最低溶解度
-
评估具有最难清洁特性的产品
-
用擦拭法最难清洁的设备位置
警告信内容详见: 详情链接
-
工艺验证:
Laboratoires Clarins警告信
公司未能验证生产工艺以及确认用于生产药品的设备。 具体而言,没用进行工艺确认试验,也没有严格的持续性计划用于监控工艺控制,以确保稳定的生产运行和一致的药品质量。
FDA要求:
-
提供确保整个产品生命周期处于控制状态的验证计划。 包括为每种药品执行适当的工艺性能确认(PPQ)的计划表。 描述用于监控批间差异的程序,以确保持续的控制状态。
-
工艺性能方案和试验,评估生产设备和工艺是否可靠。 包括但不限于:确定生产参数是否始终如一,以及工艺是否能够重复生产符合其质量属性的产品。
警告信内容详见: 详情链接
-
微生物实验室数据完整性:
Hospira Healthcare India Pvt. Ltd.警告信:
在对实验室的现场检查中,检查员观察了与无菌生产线相关的人员、环境监测培养皿上有微生物生长。 在评估实验室记录期间,发现员工记录这些培养皿“无”生长。 同一天,在3个样本上也观察到记录的微生物试验结果值明显较低。 在检查过程中,员工还确认目前的实验室记录未反映实际微生物结果。
FDA要求:
-
关于最终的整改报告,以确保无菌活动得到完全控制。 参与这些活动的人员也应完全重新获得资质。
-
关于QC实验室进行最终整改报告,以改进检查期间观察到的不适当的规程、培训和cGMP记录。
-
对包括水系统和环境监测在内的所有微生物实验室数据的可靠性进行回顾性评估。 此评估应涉及已交付至美国但尚未超过有效期的所有批次。
警告信内容详见: 详情链接
FDA于2019年7月发布了第99-32号进口警报,包含6家中国企业。根据进口警报显示,这6家企业因拒绝FDA检查,所有产品将无需实物检查即被自动扣押。这六家企业包括:
-
湖北康正药业有限公司 ;发布日期:2019-05-06
-
江西新赣江药业有限公司 ;发布日期:2019-06-17
-
Qihe Hitech Pharmchem Co., Ltd(位于中国山东齐河);发布日期:2019-03-29
-
天津和治药业有限公司;发布日期:2019-04-19
-
Tongzhou Deqi Medical Products Factory (位于中国江苏南通);发布日期:2019-05-06
-
湖北华世通生物医药科技有限公司;发布日期:2019-04-15
FDA在2014年10月发布了《构成拖延、抵制、限制或拒绝药品检查的情况》指南。依据该指南,如果一个药品制造、加工、包装或持有所在的任何工厂、仓库或场地,以及上述工厂、仓库或场地的业主、操作者或代理拖延、抵制、或限制一场现场检查,或拒绝检查员进入或拒绝接受检查,则该药品被视为掺杂药品。
美国药典的新变化:
USP第1226篇通则《药典方法确认》修订版获批准,收录于USP42-NF27。依据修订版草案,在“确认要求”引入修订:“用户有责任在整个过程中证明对照品和样品溶液的稳定性”,即检测所需溶液的稳定性需在方法确认过程中得到评估。
药典通则-包装和分销委员会(GC-PDEC)提出对以下章节的新提议:
考虑到拟议新章节<382>的范围和行业影响,GC-PDEC提出了一个为期5年的延迟实施计划,以允许行业有足够的时间来实施<382>。 一旦<382>完全实现,<381>中的功能性测试将被省略。
详情链接
2019年7月,基于美国与欧盟之间的互认协议(MRA),美国完成对斯洛伐克检查机构的能力评估,正式接受斯洛伐克。至此,美国已认可所有欧盟成员国。FDA早在2017年获得欧盟认可。FDA与欧盟之间实现互认。自2017年11月1日起,EU成员国将不会重复FDA已执行过的检查。同时,预期FDA也不会重复经认可的EU药监当局已执行过的检查。在例外情况下,EU和FDA都保留权力在任何时间在另一方管辖范围内执行检查。互认初期,EU和FDA将关注于在各自对应的管辖范围内所实行的检查。但是,EU和FDA也可以选择依赖于经过认可的药监当局,对位于其对应管辖范围以外的生产场地签发的检查报告。参见MRA的GMP部分附录第3(1)条。
详情链接
MRA协议内容详见:详情链接
EU
基于2016年出版的《“内包装灭菌工艺需要什么数据”问与答》 欧洲药品管理局(EMA)于3月发布了新的《药品、活性物质、辅料和内包装容器灭菌指南》。该指南将于2019年10月1日起生效。该指导原则包括引言、范围、法律依据、总体要求(包括对无菌药品和无菌组分的生产要求、蒸汽灭菌、干热灭菌、电离辐射灭菌、气体灭菌、除菌过滤、无菌生产工艺)、无菌活性物质和无菌辅料以及无菌包装材料生产质量管理原则和指南、灭菌方法选择、灭菌方法决策树和有关名词术语、参考文献。
根据该指南,下列关于包装组件灭菌的详细信息应包含在新上市许可申请或变更申请的注册资料中:
-
灭菌方法及灭菌循环
-
如果灭菌循环没有使用《欧洲药典》中规定的参考条件,对灭菌循环的验证
-
进行灭菌场地的名称和地址,以及场地GMP认证的详细信息(如有)。
无菌容器应尽可能在灭菌前进行包装。 当不能通过加热进行最终灭菌时,可以考虑应用除菌过滤和/或无菌工艺等替代方法。然而,使用热不稳定的容器不能成为不使用最终灭菌工艺的唯一原因,应对可替代材料进行调查。因此,应包括关于开发可进行最终灭菌的容器所做努力的讨论和全面彻底风险评估。
指南内容详见:详情链接
EDQM 于2019年6月 更新了关于沙坦类CEP申请审评和后续步骤的最新情况。
EDQM现已完成对含有四唑环结构的沙坦类药物绝大多数CEP注册资料的审查和更新。2019年4月2日,欧盟委员会发布了对含有缬沙坦、坎地沙坦、厄贝沙坦、氯沙坦和奥美沙坦的药物具有法律约束力的最终决定。本决定所涉及的5中沙坦类均包含在《欧洲药典》各自的各论中,本决定规定了2年过渡期,在此期间对NDMA( N-亚硝基二甲胺)和NDEA(N-亚硝基二乙胺)进行强制性检测。
EDQM很快将与CEP持有人联系,并要求他们将各自的CEP注册资料与修订后的专论相对应。EDQM将修订相关的CEP以包括对NDMA和NDEA的限制,无论它们是否存在。 这项工作将于2020年1月完成。
过渡期结束后(自2021年4月开始)带有四唑环的沙坦不得含有可量化水平的NDMA和NDEA (相当于低于0.03 ppm)。CEP持有人可决定立即对NDMA和NDEA申请低于0.03 ppm的限值,以避免其CEP在未来进行新的修订。过渡时期结束时,不符合这些要求的CEP注册资料必须再次进行相应更新。要求沙坦类制造商尽快验证是否有必要改变其制造工艺和/或控制策略以满足这些要求,在这种情况下,必须提交适当的修订请求和提供相关支持数据。
详情链接
欧盟Eudra GMDP于2019年3月发布关于浙江海正药业台州工厂(2家)的GMP不符合报告。在检查期间发现了25条缺陷,其中2条关键缺陷和5条主要缺陷。

关键缺陷:
一是由于在多产品设施中对非细胞毒性、细胞毒性、危险性和强毒性物质的处理不足(即主要是位于岩头厂区的Y36、Y37、Y38、Y39幢和东厂的E08号楼,还有其他建筑物也可能造成一些风险,如岩头厂区的Y20、Y33、Y35、Y50幢);
二是这些设施中的交叉污染风险没有得到适当的识别和缓解。主要缺陷主要涉及清洁验证、纯化水监测、暂存时间验证、溶剂回收等方面。
建议采取的措施:
-
不应授予此生产商原料药的任何新的/正在进行的销售许可或变更申请;
-
建议提交一份变更申请来引入原料药的可替代生产商;
-
由于2016年的非符合性申明,此厂区的活动已在欧盟市场暂停,且仅生产关键的原料药并进口至欧盟,因此目前不建议召回该厂区所生产的原料药,除非检测结果不符合质量标准;
-
建议禁止供应上述原料药,除非没有替代供应商且存在短缺风险。
其他评价:要求上市许可持有人联系相关国家主管当局,以核实所涉及原料药是否被认为是关键的。对于这些原料药,没有替代供应商,并且有原料短缺的风险,将不在不符合声明的范围之内。
详情链接
EDQM于2019年4月发布了2006-2018年原料药(API)检查及缺陷趋势汇总报告。本文件是对EDQM在2006年至2018年间之间进行的API检查数据的回顾。数据包括:
-
检查的场地;
-
首次检查以及复查;
-
检查结论;
-
检查缺陷对应欧盟GMP第二部分的分布情况及关键性;
-
缺陷出现的频率;
-
数据完整性问题。
从颁布的数据来看,在2018年,中国超越印度,成为EDQM检查最多的国家。
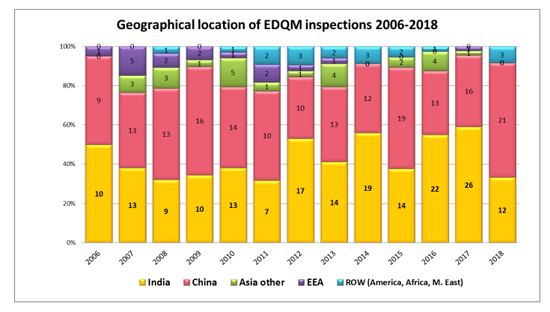
从缺陷分布来看,由质量相关因素引起的缺陷占据首位(36%),其次是厂房和设施(26%);物料管理、仓储、发运和包装(14%);实验室控制(13%);生产与工艺过程控制、物料的拒绝与拒收(7%);与CEP 注册资料一致性、《欧洲药典》符合性问题(4%)。
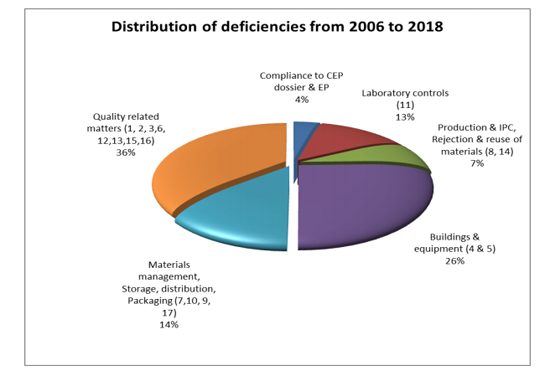
报告详细内容详见: 详情链接
截止2019年7月,2019年上半年共有20家中国药企通过欧盟GMP检查。2019年是欧盟实施(EU)2016/161《防伪指令》的第一年,处方药制造企业需要对药品实施序列化可追溯系统和防篡改装置(UI+ATD)。下列药企已顺利通过检查,并获得欧盟颁发的GMP证书:
|
公司名称
|
城市
|
检查结束日期
|
|
山东新华制药有限公司
|
淄博
|
2019/6/4
|
|
海南先声药业有限公司
|
海口
|
2019/5/10
|
|
浙江普利药业有限公司
|
杭州
|
2019/5/8
|
|
辉凌制药(中国)有限公司
|
广东省中山市
|
2019/4/11
|
|
上海恒瑞医药有限公司
|
上海
|
2019/3/28
|
|
北京费森尤斯卡比医药有限公司
|
北京
|
2019/3/27
|
|
耶赛明(南通)保健食品有限公司
|
江苏省
|
2019/3/22
|
|
上海合全药业有限公司
|
上海
|
2019/3/22
|
|
江阴技源药业有限公司
|
江苏省
|
2019/3/21
|
|
福建省福抗药业股份有限公司
|
福建
|
2019/3/21
|
|
阿斯利康制药有限公司
|
江苏无锡
|
2019/3/15
|
|
重庆伊诺生化制品有限公司
|
重庆
|
2019/3/7
|
|
联化科技股份有限公司
|
台州
|
2019/3/7
|
|
石药集团中诺药业(石家庄)有限公司
|
石家庄
|
2019/3/1
|
|
立国制药有限公司
|
广东省河源市紫金县蓝塘镇
|
2019/1/30
|
|
药明生物技术有限公司
|
无锡
|
2019/1/25
|
|
丽珠集团福州福兴医药有限公司
|
福州
|
2019/1/25
|
|
上海药明生物技术有限公司
|
上海
|
2019/1/18
|
|
江苏万邦生化医药股份有限公司
|
徐州
|
2019/1/17
|
|
齐鲁安替(临邑)制药有限公司
|
山东省
|
2019/1/16
|
EMA在其4月更新发布的《欧洲药品管理局对集中审评的用户获取许可后的建议》,旨在为集中授权的医药产品的上市许可持有者,提供授权许可后变更分类的建议。更新版指南新增或修订了21个问答,涵盖行政变更、质量变更、(非)临床变更和编辑性变更等。在第7部分关于变更分类的问答中,部分翻译如下:
-
7.2.9. 生产场地、厂房和房间的哪些变更可以由公司质量保证体系 (GMP) 覆盖?
答:如果模块3不受影响, 除3.2.A.1 节 (生物医药产品))外, 下文列出的变更 (非详尽清单) 将由公司的质量管理体系覆盖, 不需要上市许可变更:
-
生产活动从一栋建筑转移到同一许可地点的另一栋建筑
-
生产活动从同一许可建筑的一个房间转移到另一个房间
-
QC 活动从一栋建筑转移到同一许可地点的另一栋建筑
-
在已许可的房间、建筑、生产场地上新增一条与已批准的完全相同的生产线
-
已许可建筑中新增隔离器
-
在已许可的建筑内新增培养基或缓冲液配制间
-
已许可生产基地的布局变化
如果由于上述任何变更, 导致对模块 3 (生物医药产品3.2.A.1 节除外) 的任何变更, 如生产工厂地址细节的变更、生产工艺的变更、批量变更等, MAH 应提交适当的变更。
-
7.2.10 生产过程中使用的设备变更,公司质量保证体系 (GMP) 涵盖哪些变更?
答:只要新设备与目前使用的设备相当, 并在批准的工艺参数范围内运行, 则变更由公司的质量保证体系覆盖。如果引入新设备对模块3中登记的工艺和细节有任何影响 (生物医药产品3.2.A.1 节除外),则MAH应提交相应的变更。
查看详情
欧盟更新边界类医疗器械的划分指南。何种程度的医疗设备被认为是欧洲监管下的医疗器械? 该指南对临界产品的分类提供指导。边界产品指从一开始就不清楚是属于医疗设备、体外诊断医疗设备还是植入式医疗设备。 或者,从定义上属于医疗器械,但在使用范围上被排除在监管法规之外。 该指南列举了具体的边界类产品实例,为分类提供指导。
指南内容详见:
详情链接
第10版《欧洲药典》更新
-
更新第三章,增加3.3包装章节内容包含人体血液和血液成分的容器,以及制造过程中使用的材料;输液器和制造过程中使用的材料;注射器。
-
增加5.25章节《 过程分析技术》(PAT)
-
更新通则第2.2.25 篇《紫外与可见光吸收光谱》,该章节由EP通用方法工作组紫外专家们进行修订。应用上不仅包含比色池测量,还将其与色谱系统联合,或作为PAT的一部分,以及其他更为集成的应用。除了扩展应用范围外,该章节其它主要变化包括:
-
优化文章结构;
-
设备和其操作有更为详细的描述;
-
识别紫外-可见吸收分光光度法不同用途的特殊性,并对每种方法提出建议和要求;
-
增加系统适用性测试部分;
-
引入烟酸作为重铬酸钾的替代品,以控制吸光度准确度;
-
设备性能控制全面明确的要求(波长精度、吸光准确度、光度线性、散光限、分辨率/光谱带宽),保留了在选择对照品时的灵活性。
-
其他:
-
修订计算有关物质检验中百分比含量时使用的措辞,有关物质已被修改为指定物质的全名(即包括抗衡离子、水合物等);
-
修订使用五氧化二磷的检测,主要是对干燥失重检测进行了改进,以避免因其毒性而使用;
-
修订用于描述GC固定相试剂的名称和描述;
-
替换“防篡改(Tamper-proof)”为“显窃启(Tamper-evident),参考2.8.23章;
-
使用具有非标准筛尺寸的粉末进行显微镜检查时,添加草药的显微镜检查;
-
从“粗粉(coarse powder)”和“粗粉草药(coarsely powdered herbal drug)”中删除了“粗(coarse, coarsely)”的描述。(2.9.12。筛分试验);
-
2.8.16提取物的干燥残留和3.8.17 提取物干燥失重的检测结果,以%表示,不再以% m/m表示。
第10版《欧洲药典》将于2019年7月发布,并将在未来3年进行8次增补。 第10版的新修订文本将于2020年1月1日正式生效。
详情链接
WHO
世卫公布了关于使用非蒸馏方法生产注射用水(WFI)的指南草案,公开征集意见。已适应欧洲药典对注射用水的修订。该指南包含以下章节:
-
介绍
-
范围
-
专论:生产商应有WHI质量标准,所生产的WFI质量应满足药典要求。
-
生命周期方法:要求WFI的生产应符合“良好实践”,并且涉及源水处理、生产、储存和分配以及控制。
-
风险评估:应选择生产WFI的“适当”方法,并应包括基于生命周期考量相关的风险。 控制措施应更加具体。控制应确保没有水污染的风险,没有死角以及使用在线监测等。
-
控制策略:要求对WFI生产进行确认和验证。对于生产WFI进水(feed water)的处理提出要求, 应确保去除无机和有机化学物质、以及颗粒和微生物杂质。 但是这些处理,不应对水处理组分的质量产生任何负面影响。在讨论可能的处理技术时,提到了去离子,超滤,除垢,脱气,UV处理,其可以与双程反渗透结合使用。
-
WFI生产中的良好做法:讨论用于生产WFI的进水的质量。至少应符合WHO或其他国家饮用水或纯化水的质量要求。 要求对电导率和TOC进行监测和趋势分析,还需要对反渗透膜进行监测。
指南草案内容详见:
详情链接
WHO在2019年5月颁布的第1019号技术报告附件二中通过了《非无菌产品空调系统指南第二部分》,部分内容解读如下:
-
风险评估:应对暖通空调系统进行风险评估,风险评估可选择失效模式、影响分析(FMEA)和故障树分析(FTA)。例如,暖通空调系统中的一个或多个空调机组失效的影响;除尘系统失效;对过滤器、加热管、冷却管、风扇等空调系统组件进行评估,并确定和实施适当的控制措施。考虑的因素包括但不限于:空调机组可能产生的失效、季节变化、原料和产品的性质和种类、人员数量和交叉污染的风险。
-
对于称量区域,建议设置称量前室、人员气闸室、物料气闸室、带称量罩的称量室、称量后室、清洗区和废料传出设施。
-
应特别注意门的设计,因为门和地板之间的缝隙,门朝低压区打开和推拉门都可能导致不同区域间压差的变化。
-
关于空调关停操作,如果制造商决定在特定的时间段(如夜间、周末)或长时间使用节能模式或关闭某些空调机组,则应注意确保原辅料和产品不受影响。在这种情况下,决定、程序和记录应有充分的文件记录。包括风险评估、标准操作规程(SOP)、空气处理设备启动和关闭顺序的规程和记录以及验证。
-
对于再循环系统,应在验证期间确认其新风比。
-
关于换气次数,推荐值为6-20次/小时,自净时间为15-20分钟
-
关于压差,建议为5-20pa之间
报告内容详见:
详情链接
WHO计划修订其《良好贮藏及发运规范》指南。 在第五十三届药品标准专家委员会(ECSPP; 2018年10月)期间,专家委员会建议将《药品的良好贮藏和良好发运规范》以及《良好发运渠道指南》的要素合并为一份文件。 该指南包含参与贸易、储存和分销的各方,例如:
-
制造商
-
批发商
-
经纪人和贸易商
-
供应商
-
经销商
-
物流提供商、运输公司和货运代理。
关于重新包装和重新贴签:
-
不建议对物料和产品进行重新包装和重新贴签,如确实有,则只能由经适当授权的机构按照适用的国家、区域和国际要求并按照 GMP 执行。
-
应当制定程序对原包装进行受控处理,防止再次使用。
关于运输过程的控制,要求如有可能,应考虑使用全球定位技术(GPS)电子追踪装置和发动机熄火按钮控制以加强车辆与产品的安全性和可追溯性。 在集装箱内使用干冰时应特别注意安全问题和可能对产品造成的不良影响
征求意见稿已在当前项目下的WHO药品网站上公布。
指南草案内容详见:
详情链接
WHO于2019年5月发布新的指南草案《GMP中的环境方面:制造企业和检查员关于防止抗生素耐药性方面的考虑点》,拟将EHS纳入GMP管理。WHO 表示:“鉴于对药物生产下游流程缺乏控制将最终导致疗效丧失,不能仅关注与药品质量直接相关的 GMP 领域。”
这份 24 页的文件旨在增强人们对管理抗微生物药生产废物和废水相关 GMP 的意识。考虑所有 GMP 实施方面,并关注极为重要的抗菌药,以建立和执行废物和废水安全处理要求。草案还提出了控制和减少生产过程中抗微生物药和化学品环境污染的方法。
WHO 预认证小组检查部门还计划明年启动一项试点,检查那些极为重要的抗微生物原料药和成品制剂的生产商是否符合 WHO 关于含有有害物质药品的 GMP 附录。该指南部分要求如下:
-
GMP 检查机构应对检查员如何检查废物和废水管理过程开展培训 , 并且应指导他们在检查生产极为重要的抗微生物药场地期间,检查废物和废水管理过程。GMP 检查机构还应与负责执行环境保护标准的相关方密切合作。
-
WHO 建议将抗微生物药包含在 TRS957 附录3《含有害物质的药品》中,该附录介绍了在某些激素、类固醇和细胞毒素生产中废水处理方面的 GMP。附录要求进行废水分析,有害物质转化为非有害废物的月度报告及记录。
-
生产商应建立抗微生物药废物处理的行业标准。
-
所有 WHO 成员国都应制定 AMR 行动计划,并加强水和废水要求的立法及强制执行。
-
废水处理人员应接受抗微生物药净化培训 , 并应有监测废水中抗微生物药的计划。
详情链接
WHO 预认证
WHO于2019年5月更新了通过WHO预认证的原料药清单。拥有良好质量的API对于生产优质制剂至关重要。WHO的API预认证通过确定制剂生产商(FPP)使用的API来促进优质药品的生产。
API预认证包括一个综合评估程序,该程序包含两个部分:评估API主文件(APIMF)来确认是否符合WHO规范和标准。以及评估API制造地点以确认是否符合WHO良好生产规范要求。
在最新更新的清单里,2019年上半年(截止7月23),新增13种 API通过预认证,其中有4种来自国内企业。分别是广西桂林南药股份有限公司的青蒿琥酯,上海迪赛诺化学制药有限公司的洛匹那韦和阿扎那韦(硫酸盐),以及浙江江北药业有限公司的地瑞那韦(乙醇盐)。
详情链接
WHO 于2019年7月发布对“非昔硝唑、左氧氟沙星、利奈唑胺、莫西沙星、奥司他韦、利巴韦林以及扎那米韦”7种药物的生物等效性试验指南。为一致性评价提供方法指导。
详情链接
WHO于2019年6月发布产品召回 ,对Macleods制药有限公司生产的莫西沙星100mg分散片(TB342):批次BMC5801A, 进行召回。
2019年5月28日,Macleods制药有限公司通知WHO预认证小组PQT:已获得WHO PQT预认证产品的检测结果不合格(药品,TB342盐酸莫西沙星分散片100 mg,批号BMC5801A )。
TB342盐酸莫西沙星分散片100mg商品批中的一批(即批号BMC5801A),于2018年9月生产(有效期:2020年8月,Blister,Alu / Alu 10x10),在30℃/ 75%RH(实时)条件下储存时,溶出试验结果未能满足NLT 80(Q)在S3阶段15分钟的验收标准。
2019年6月1日,Macleods 制药有限公司发起了该产品的自愿召回。该公司进行了健康危害评估,确定使用该产品不存在安全问题,并且莫西沙星分散片100 mg的治疗活性不太可能受到影响。
详情链接
召回报告详见:点我
其他组织及机构
于2019年2月发布新技术报告TR81 《基于细胞治疗的控制策略》,该技术报告侧重于开发适用于细胞治疗基于风险的控制策略,以降低产生不良质量产品的风险。
详情链接
APIC
2019年3月更新了对ICH Q7实施“HOW TO DO”(如何理解实施ICH Q7)文件。更新了对ICH Q7第三章“人员”、第五章“物料管理”以及第十七章“代理商、经纪人、贸易商、经销商、重新包装商及重新贴标商”的解读。
详情链接
2019年3月发布了《基于风险的实用数据完整性管理指南》第一版。该指南强调了创建和维护支持数据完整性的工作环境和组织文化的重要性。 公司应建立数据治理计划,以解决技术、程序和行为方面的问题,以确保对数据质量和完整性的信心。
详情链接
2019年5月28日,由Intertek天祥集团与湖北省标准化研究院联合承办的第二十一届中国制药行业论坛(China Pharmaceutical Forum,下称“CPF”)在湖北武汉完满落幕。中国制药论坛-CPF创立于2010年,迄今已经在上海、南京、杭州、重庆、北京、山东等地举办了20期。现任组委会的5家成员单位为美国食品药品管理局(FDA)中国办公室、中国医药保健品进出口商会、上海勃林格殷格翰药业,常州四药和Intertek天祥集团。CPF 旨在为医药行业和药政官方搭建一个技术交流平台,解读最新政策法规和分享实践经验,助力医药行业发展。
本次论坛分别从监管、迎检以及审计三个方面,特邀中国医药保健品进出口商会、湖北省药监局、美国FDA、药明生物等机构嘉宾以及业界专家学者,为与会者带来精彩的政策解读和经验分享。论坛由Intertek副总裁薛艳女士主持,中国医保商会何春红主任分享了医药保健品进出口最新的趋势与数据。由前FDA中国办公室副主任辛强博士主持的论坛嘉宾讨论环节,各位专家为华中地区医药企业更好地“走出去”,同时把国外新技术“引进来”提出了建设性的建议。本次论坛座无虚席,研讨内容获得参会人员的一致好评。
详情链接
联系我们
联系人:韩小姐 Lara Han
电话:021 5339 7970
手机:176 6133 1359
E-mail:lara.han@intertek.com
|


