ICH
ICH Q12指南被采用,进入第四阶段
2019年11月16至20日,国际协调理事会(ICH)在新加坡举行会议。达成的关键里程碑事件是采用正式通过了ICH Q12新指南《医药产品生命周期管理的技术和监管考量》(ICH进程的第四步) 。包括两个附件在内的ICH Q12新指南是ICH质量指南Q8至Q11的补充。 ICH表示,Q12旨在“促进制药行业的创新和持续改进,并且加强质量保证和产品的可靠供应,包括全球供应链调整的前摄性规划”。
为确保采用标准化的方式,该指南定义了批准后“化学、制造与控制(CMC)”变更分类、既定条件(Established Conditions ,EC)、批准后变更管理方案(Post-Approval Change Management Protocols ,PACMP)、产品生命周期管理(Product Lifecycle Management ,PLCM)概念的。该指南特别强调了注册审评(Regulatory Assessment)和GMP检查之间的关系。
在ICH网站上发布Q12指南及其附件最终版之后,下一个阶段将是在ICH各地区实施ICH Q12。但是,必须全面修订地方法规(特别是在欧盟,例如,欧盟变更法规),Q12的概念才能得到全面贯彻。在美国,ICH Q12指南与既定法律框架完全兼容。
ICH Q12指南和附件可在ICH网站下载:详情链接
ICH指南制定进展
11 月 4 日,加拿大卫生部和美国国家局(FDA) 在渥太华举行了 ICH 联合公众咨询会,参会官员在会上简要介绍了20多份 ICH 指南的进展。六个月前,即今年6月,在ICH大会会议于阿姆斯特丹召开前,美国国家局 与加拿大卫生部曾一同主办前次联合公众咨询会。
加拿大卫生部治疗产品部心脏病、过敏和神经科学局局长利奥·布锡莱勒 (Leo Bouthillier )介绍了 14 个正在进行的主题和工作领域。
一些重大主题包括:
|
指南/工作项目
|
主题
|
状态
|
|
E17 培训材料
|
多区域临床试验
|
在ICH 网站7个培训模块可用
|
|
M7(R2)
|
评估和控制药物中DNA反应性(致突变)杂质以限制潜在致癌风险
|
M7 附录——专论草案预计2020年发布
M7 问与答文件预计2019年底发布
|
|
M10
|
生物分析方法验证
|
预计2020年11月出最终指南
|
|
Q3C(R8)
|
残留溶剂指南的维护
|
预计在2019年底之前出针对3个新溶剂的指南草案
|
|
Q3D(R1/R2)
|
元素杂质指南的维护
|
预计2019年底之前出附录草案
|
|
Q13
|
原料药和制剂的连续制造
|
预计2020年6月出第二步草案
|
|
Q2(R2)/Q14
|
分析规程开发和Q2(R1)分析规程验证的修订
|
预计2020年6月出指南草案
未来可能合并两指南
|
|
Q11 问与答培训材料
|
起始物料的选择与论证
|
最近已在ICH网站发布
|
|
M8 专家工作组
|
电子通用技术文件(eCTD)
|
为eCTDv4 更新材料和文件
|
来源:详情链接
美国国家局(FDA)
2019财年(2018.10-2019.09)警告信趋势
在2019财年,美国国家局共向全球成品药制造企业签发了81封警告信,达5年之最(自2018年10月至2019年9月),签发给配药或药房制剂的警告信不在此统计之列相比之下,签发给原料药I制造企业的警告信在2017财年达到峰值之后,在2019财年数量已降至5年前水平。
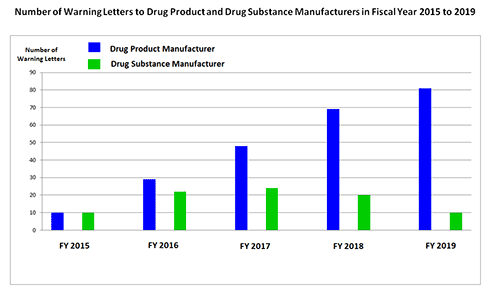
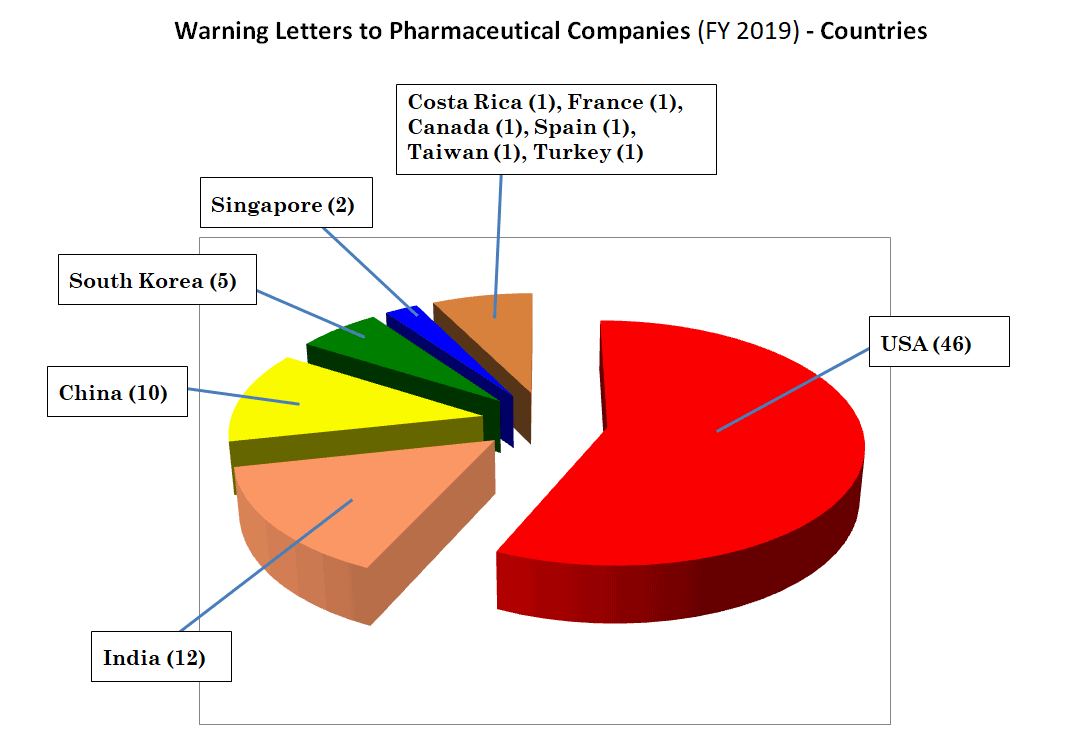
如下图所示,这81封警告信中,46封发给美国企业、11封发给中国企业(含1家台湾企业)、12封发给印度企业。这表明cGMP合规不仅是中国和印度企业面临的挑战,美国本土药企也一样面临挑战。
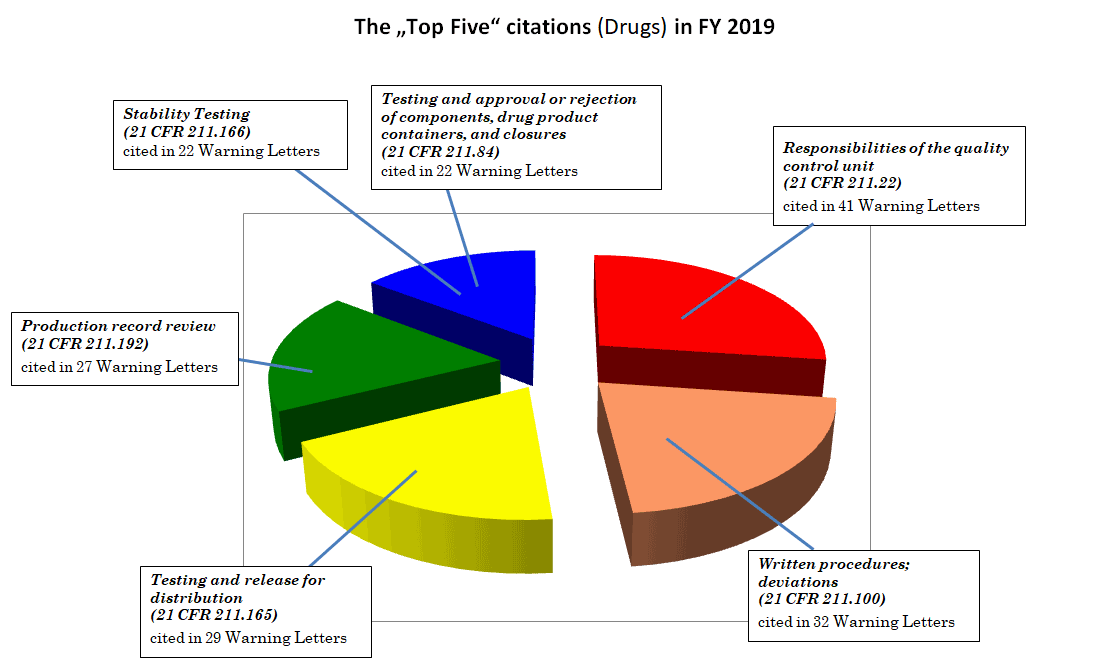
与前几年一样,对过去12个月中警告信内容的分析显示出类似情况:最常见的GMP违规行为涉及每个制药公司理应遵守的基本要求。下图说明了警告信中缺陷项引用频率最高的美国联邦法规第211部分(cGMP)中的章节,前五名分别为:
-
21 CFR 第22节,质量控制部门职责 (41封警告信引用)
-
21 CFR 第100 节,书面规程;偏差(32封警告信引用)
-
21 CFR第165节,检验与放行销售(29封警告信引用)
-
21 CFR 第192 节,生产记录审核(27封警告信引用)
-
21 CFR第166节, 稳定性试验(22封警告信引用)
-
21 CFR 第84节,组分、药品容器和密封构件的检验、批准或拒收(22封警告信引用)
以下是引用违反第211.165节检验与放行销售以及第211.166节稳定性试验的一些实例:
你公司未制定检验方法,并且未将检验方法的准确性,灵敏性,专属性和重现性文件化(21 CFR 211.165(e))。
在放行销售之前,你公司未验证用于检验制剂中活性成分含量的分析方法。对于同一个活性成分分析,你们的笔记本和高效液相色谱报告包含不同的仪器参数。
在放行之前,你公司没有对每批药品进行适当的实验室检验,以确保符合成品最终质量标准,包括每种活性成分的特性和规格(21 CFR 211.165(a))。
你公司放行了非处方(药,但未执行任何最终成品检验,包括但不限于活性成分的特性和规格……
……你们检验了用于……的…批某原料药,所有结果均报告为合格。但是,在国家局检查中……,我们要求在我们观察下复测相同的批次,所有复测结果均为超标结果。
你公司未建立并遵守适当的书面检验计划以评估药品的稳定性特征,并且以使用此类稳定性检验结果来确定适当的贮藏条件和有效期(21 CFR 211.166(a))。
你公司未实施长期室温稳定性试验来支持你们非处方药的有效期。
你公司没有一个适当的稳定性试验计划来证明你们非处方药的化学特性在标示的整个有效期内保持可接受。例如,你们的稳定性试验计划未涵盖非处方药中活性成分的含量测定。
你们缺乏适当的稳定性数据来支持标示的两年有效期……。具体来说,你们没有按照在用于支持有效期的加速稳定性计划中指定的时间间隔来检验含量,你们也没有完整的长期稳定性数据。因此,你们没有适当的数据来支持你们产品在标示的有效期内的稳定性。
参考资料:
详情链接1
详情链接2
美国药品评价与研究中心新药计划2019年进展
12 月 3至4日,在美国弗吉尼亚州阿灵顿市召开了2019 年美国国家局与医疗保险和医疗补助服务中心(FDA/CMS)峰会。今年会议讨论重点包括:
-
新药审评流程的效率:处方药用户付费法案(PDUFA)第六期历史回顾和进行中的变化
-
2019年新药活动:获批情况、工作量、国际比较,以及对2019年新分子实体和生物制品许可申请类别的分析
-
研发阶段活动:研究性新药申请(IND)数量、突破性计划、实现工作量以及处方药用户付费法案第六期的持续变化
-
展望2020年:新药监管计划现代化工作
美国国家局新药办公室运营副主任库什布·夏尔马(Khushboo Sharma)介绍了 2019 年美国新药审批情况,包括处方药用户付费法案第六期实施情况以及 2019 年的新药批准工作回顾,并展望了 2020 年将实施的新药监管现代化工作。
2019 财年,药品评价和研究中心批准了 45 个新分子实体,包括 23 个罕见病用药。45 个获批新药中有 32 个为优先审评。约有一半的新分子实体用于治疗罕见病。具体为:
-
一半以上罕见病用药(12/23)用于治疗癌症或癌症成像
-
4个神经科药物,用于遗传性甲状腺素介导的淀粉样变性病的多发性神经病(LEMS),以及两个用于成年发作性睡病患者的日间嗜睡(EDS)的药物
-
3个罕见病用药用于遗传条件:遗传性甲状腺素转运蛋白介导的淀粉样变性多发性神经病,原发性噬血细胞淋巴组织细胞增生症(HLH),腺苷脱氨酶严重合并免疫缺陷病(ADA-SCID)
值得注意的是,2019年11月我国百济神州开发的布鲁顿酪氨酸激酶抑制剂泽布替尼(Brukinsa)获得美国国家局加速批准,用于治疗套细胞淋巴瘤。这是获得美国批准的首个中国原研抗癌药。这一突破不但体现了中国医药公司创新药研发能力的进步,也是中国监管机构进行改革,与国际标准接轨的成果。在支持泽布替尼获批的两项临床试验中,在中国进行的名为BGB-3111-206的多中心2期临床试验数据与全球性2期临床试验数据一起支持泽布替尼的加速获批。
完整报告详见: 详情链接
2019年批准新药数据详见FDA官网: 详情链接
新增的指南
? 行业指南:药物主文件(DMF) 草案(2019-10-18)
该指南代表美国国家局当前对药物主文件(DMF)的观点。药物主文件F是向美国国家局提交的申报资料,可用于提交关于人用药制造、加工、包装和贮藏所用设施、工艺或物品的详细机密信息。药物主文件亦可包括其它类型的资料(例如,毒性资料、共享系统REMS(风险评价和减缓策略))。药物主文件持有人可授权一个或多个(制剂)申请人或申办人通过引用药物主文件中包含的信息合并到制剂申请中,而无需向制剂申请人或申办人披露这些信息。药物主文件的提交由持有人自行决定,并无法律或法规要求。通常不针对非专有物料提交。通常美国国家局不单独审评或批准药物主文件,相反,仅在审评引用该药物主文件的制剂申请时才关联审评药物主文件的技术内容。
药物主文件可用于支持(但并非替代)由美国国家局审评的制剂申请。该指南重点关注向药品评价与研究中心(CDER)和生物制品评价与研究中心(CBER)提交的以下申报资料:
-
依据联邦法规第二十一篇(21 CFR )第420条提交的药物主文件,用于支持依据《联邦食品、药品与化妆品法案》提交的新药申请、简略新药申请和研究性新药申请
-
依据联邦法规第二十一篇 第51(a)条提交的药物主文件和其它主文件,用于支持依据《公共卫生服务法案》提交的生物制品许可申请
另外,药物主文件中所包含的信息一般可在医疗器械(例如,上市前批准)和兽药(例如新兽药申请)的上市前提交中引用。虽然该指南重点在于上述向CDER和CBER提交的申请资料,但美国国家局相信该指南的内容将有助于其它主文件持有人向国家局提交符合最新要求的完整主文件。
指南全文:详情链接
《行业指南:识别向CBER和CDER提交的申请资料中的制造设施之问与答》草案(2019-10-22)
该指南意在澄清美国国家局关于应当包括在下述申请以及对这些申请类型的重新申请中的设施信息的要求:初始新药申请、简略新药申请、初始生物制品许可申请、修订、增补、“化学、制造与控制CMC”增补。
提交FDA-356h表符合申请人提交申报表的要求(21 CFR 314.50(a) 和314.94(a)(1); 601.2(a))请人。FDA-356h表既是行政信息综述,也是与申请资料相关原料药与制剂设施所有制造、包装和检验场地的位置的完整信息汇总。
该指南阐述在申请资料中包含或撤销拟注册的商业设施和研发设施、设施信息在申请资料中的适当位置,以及申请资料中应包含的设施信息类型等问题。在FDA-356h表设施信息章节涵盖恰当和完整的设施信息的申请资料,将会降低发补(Information Request ,IR)、拒绝立卷(RTF)和拒收(RTR)的频率,提高申请资料审评流程的效率。该指南阐述了初始申请和增补申请资料中所有设施信息放置位置的建议。
该指南适用于依据《公众卫生服务法案》第351条许可的生物制品许可申请(BLA)产品,包括按该类产品监管的体外诊断产品,以及依据《联邦食品、药品与化妆品法案》按照新药申请或简略新药申请上市(或待上市)的制剂。该指南适用于所有制造场地,包括依照合同履行职能的设施。
指南全文:详情链接
《行业指南:透皮和局部给药系统之产品研发和质量考量》草案 (2019-11-20)
该指南为透皮和局部给药系统(TDS)的申请人和制造企业提供有关药物开发和质量信息的建议,以包括在新药申请和简略新药申请中。 具体来说,该指南讨论了国家局当前在产品设计和药物开发、制造过程和控制以及成品控制方面的观点。 还阐述了质量与产品性能和潜在安全问题紧密相关的领域的特殊考虑,例如粘附失败以及施加的热量对药物输送的影响。
指南全文:详情链接
国家局提议撤销4件新药申请的批准
在11 月 18日,《联邦公报》(Federal Register)正式出版前,美国 国家局宣布计划撤销4 件新药申请的批准(见下表),因为 新药申请 持有人未根据法规要求(21 CFR 第314.81条)向国家局提交年度报告。国家局指出,国家局已与这些公司联系,但这些公司屡屡未能提交所要求的报告。
年度报告旨在为国家局提供有关前一年可能会影响药物安全性、有效性或标签的更多信息。年度报告还必须简要描述申请人由于新信息而已采取或打算采取的行动,例如,提交标签补充,在标签上添加警告或发起新研究。
|
申请号
|
药品
|
持有人
|
|
NDA 014217
|
MAOLATE (氯苯甘油醚) 片剂, 400 mg
|
Pan American Laboratories, LLC, 4099 Highway 190, Covington, LA 70433.
|
|
NDA 018663
|
CHYMODIACTIN (木瓜凝乳蛋白酶) 注射剂, 4,000 单位/瓶 和 10,000 单位/瓶
|
Chart Medical, Inc., c/o Renascent Medical, Inc., 9600 Great Hills Trail, Suite 150 West, Austin, TX 78759.
|
|
NDA 020530
|
IONTOCAINE (肾上腺素和利多卡因盐酸盐) 外用液,0.01 mg/ml; 2%
|
Iomed, Inc., 2441 South 3850 West, Suite A, Salt Lake City, UT 84120-9941.
|
|
NDA 021504
|
LIDOSITE 外用系统: LidoSite Patch (盐酸利多卡因和肾上腺素外用离子电渗贴片) 10%/0.1% and LidoSite 控制器
|
Vyteris, Inc., 13-01 Pollitt Dr., Fair Lawn, NJ 07410.
|
这类通告并非罕见,特别是如果公司已经停止销售产品有一段时间。
值得注意的是,其中3个产品均为“老”产品,于 1965 年至 1995 年获得批准。“Lidosite”属于较新的申请,于 2004 年获批。
来源: 详情链接
美国国家局雷尼替丁事件更新
在过去数周,国家局陆续发布了常见的胃灼烧治疗用非处方药和处方药(雷尼替丁和尼扎替丁)中已知杂质N-亚硝基二甲胺(NDMA)检出情况。国家局已发起调查,以确定这些药品中出现该杂质的原因,并为使用这些药品的患者和消费者提供信息。作为该调查的一部分,国家局已要求制造企业在实验室自行检验雷尼替丁和尼扎替丁中NDMA含量,并寄样品给国家局,由国家局科学家检验。
2019年12月04日,国家局宣布已要求雷尼替丁和尼扎替丁制造企业扩大NDMA检验以涵盖所有药品批次,检验之后方可提供给消费者。如果检验显示NDMA超过可接受日摄入限度(对于雷尼替丁,限度为96ng/天或杂质浓度0.32 ppm),则制造企业必须通知国家局,并且不得将该批次放行。
国家局将继续与企业和全球药监机构合作,确定这些药品中NDMA出现的原因,并已开发和发布了雷尼替丁中NDMA的多个检验方法。科学家已确认在一般胃内环境下雷尼替丁不会形成NDMA。但是还需要进一步调查,对雷尼替丁和尼扎替丁在人体内行为进行全面测试,目前已有对此进行研究的计划。目前已有证据证明当存在雷尼替丁或尼扎替丁时,亚硝酸的存在与体内NDMA的形成有关联。鉴于此,希望继续使用这些药品的消费者应考虑限制摄入含亚硝酸的食物,例如,加工过的肉制品、防腐剂(例如亚硝酸钠)。
消费者可考虑使用其它已批准用于同类症状的非处方药。国家局对Pepcid(法莫替丁)、Tagamet(西米替丁)、Nexium(埃索美拉唑)、Prevacid(兰索拉唑)和Prilosec(奥美拉唑)等替代药品的检验显示不含NDMA。
12月18日,国家局提醒患者和医疗专业人员格伦马克制药公司(Glenmark Pharmaceutical Inc.)自愿召回雷尼替丁片(规格150 mg、300 mg)处方药,因为它们可能含有不可接受水平NDMA。
来源: 详情链接
美国境外糖尿病药品中发现亚硝胺类杂质
美国国家局还在继续调查某些类别药品中出现称为亚硝胺的基因毒性杂质。在过去的一年半时间,在包括血管紧张素II受体拮抗剂(ARB)和雷尼替丁(善胃得)在内的几种药品中均发现少量亚硝胺杂质,例如N-亚硝基二甲胺(NDMA)。在此期间,国家局持续调查了其它药品中亚硝胺杂质情况。此工作主要确保美国公众所用药品持续符合严格的质量标准。
美国国家局了解到其它国家报告在一些二甲双胍糖尿病药品中含有低水平NDMA。根据所获得的信息,在美国境外发现的NDMA水平处于一些食物和水中自然存在的范围内。虽然美国以外的一些药监机构可能会召回一些二甲双胍药品,但这次目前暂无二甲双胍召回影响到美国市场。美国国家局正在调查美国市场的二甲双胍是否含有NDMA,以及其水平是否超出可接受日摄入限度(96 ng)。美国国家局也将与企业合作,对在美销售的二甲双胍样品进行检验,如果发现其中NDMA水平较高,将建议酌情召回。如果调查后召回二甲双胍,国家局将及时为患者和医疗专业人员提供更新信息。
二甲双胍是一种用于控制二型糖尿病患者高血糖的处方药。患者应持续服用二甲双胍以保持糖尿病受控。对于患有这类严重疾病的患者来说,如果不与医护专业人员沟通即停止服用二甲双胍,可能有危险。国家局建议处方医生在临床适合时继续使用二甲双胍,因为调查仍在进行,并且目前无以相同方式治疗的替代药品。
来源: 详情链接
欧盟
欧洲药品管理局启动无菌药品制造企业现场检查国际试点项目
12月17日,欧洲药品管理局(EMA)宣布与欧洲合作伙伴、国际合作伙伴发起一项试点项目,以加强对人用无菌药品制造企业现场检查的合作。该项目的启动基于从类似合作项目(即原料药国际现场检查项目)中获得的成功经验。
在无菌药品现场检查合作项目中,欧洲药品管理局、欧盟成员国国家局(法国国家局(ANSM)和英国国家局(MHRA))、美国国家局(FDA)、澳大利亚国家局(TGA)、加拿大卫生部(HA)、日本国家局(PMDA)和世卫(WHO)将分享对参与国境外的无菌药品制造企业的现场检查信息,并对共同关心的制造场地组织联合检查。
对于联合检查和针对待检查的场地,上述机构分享的信息包括但不限于:
-
无菌药品名称、药用活性成份名称、目标市场(如有);
-
场地主文件(SMF)和验证主计划(VMP);
-
产品质量回顾(PQR);
-
制造工艺描述(至少工艺流程图);
-
待检查的建筑物或生产线;
-
先前风险评估或场地符合性档案或文件;
-
与待检查场地相关的其他信息。
由此可见,场地主文件、验证主计划、产品质量回顾是无菌制药企业应对国外检查应重点准备的三大核心文件。
参与机构列表
|
澳大利亚
|
药品监管局(TGA)
|
|
加拿大
|
卫生部(HC)
|
|
欧洲
|
欧洲药品管理局
|
|
法国
|
国家卫生与药品安全局(ANSM)
|
|
日本
|
医疗器械与药品局(PMDA)
|
|
英国
|
药品与医疗产品监管局(MHRA)
|
|
美国
|
食品与药品管理局(FDA)
|
|
世界卫生组织(WHO)
|
通过各参与方药监机构的相互依赖,已证明国际检查合作有利于改进对制造企业的监管,最大限度利用全球检查资源,减少重复检查,以及增加全球被检查场地的覆盖率。已发布的《人用无菌药品制造企业药品生产质量管理规范 检查国际合作试点计划》规定了该项合作的目标、范围和一般原则。
GMP 要求列表
|
检查机构
|
GMP要求
|
|
澳大利亚国家局(TGA)
|
《欧洲药典》或《英国药典》或《美国药典》
国际药品认证合作组织(PIC/S)药品生产质量管理规范(GMP)
|
|
加拿大卫生部(HC)
|
食品与药品法规
食品与药品法案
|
|
法国国家局(ANSM)
|
《欧洲药典》
《欧洲药品法》第四卷
|
|
日本国家局(PMDA)
|
药品与医疗器械法案
药品生产质量管理规范部长令
《日本药典》
国际药品认证合作组织(PIC/S)指南
|
|
英国国家局(MHRA)
|
《欧洲药典》
《欧洲药品法》第四卷
|
|
欧洲药品管理局(EMA)
|
《欧洲药典》
《欧洲药品法》第四卷
|
|
世界卫生组织(WHO)
|
《国际药典》
世卫技术报告系列(主要的GMP文本:第986号技术报告附件二(2014年版)及支持性指南
|
|
美国国家局(FDA)
|
《食品、药品与化妆品法案》
《公共卫生服务法案》
《美国联邦规章典》(CFR)
第7356.002A/M号《符合性程序》
国际药品认证合作组织(PIC/S)指南
|
上表显示,对目的国药典要求的遵守是必备条件之一。
该项目涵盖的产品范围为化学来源和某些治疗用生物技术衍生产品(例如,单克隆抗体和重组蛋白)的人用无菌药品。疫苗、细胞和基因疗法以及血浆衍生药物目前不在该项目范围内。试点项目将持续至少2年,之后各参与方将对项目进行评估,决定下一步合作措施。
文件全文: 详情链接
欧盟GMP不符合通报
2019年11月,欧盟药品检查数据库(EudraGMDP)发布了华北制药河北华民药业有限公司的不符合通报,不符合情况如下:
根据欧洲药品质量管理局(EDQM)检查计划,于2019年9月2日至6日执行了此次检查。检查范围为无菌头孢噻肟钠(CEP 2014-197)。检查共发现31条缺陷,其中2条关键缺陷,5条重大缺陷。另有一条缺陷被认为与注册资料(CEP 2014-197/头孢噻肟钠,无菌)不一致。
关键缺陷与无菌制造操作有关(103车间,C线):无菌制造设施中的微生物环境监测和无菌消毒剂的配制、灭菌和使用。
查出重大缺陷的领域是:质量保证(上次EDQM检查后整改与预防措施的落实);文件记录控制;无菌内包材的准备和处理;电子记录和签名;制造区域和设备。

参与检查的欧盟成员国药监局(NCA)采取或提议的措施:
-
上市许可变更要求:不得将此不符合通报中列出的无菌原料药制造企业列入任何新的和正在进行的上市许可申请或变更申请中。建议提交变更申请,使用替代制造企业。
-
已放行批次的召回:由相关国家监管机构与上市许可持有人评价后决定是否启动药品召回。
-
禁止供应:建议禁止无菌头孢噻肟钠、无菌头孢呋辛钠和无菌头孢曲松钠的供应,除非没有替代供应商,并且存在短缺风险。
-
暂停或撤销欧洲药典适用性证书(CEP)(由EDQM 采取措施):暂停 以下CEP:CEP 2014-197/头孢噻肟钠,无菌;CEP 2014-021/头孢呋辛钠,无菌。
-
其他:建议对无菌头孢噻肟钠、无菌头孢呋辛钠和无菌头孢曲松钠采取同样的监管措施,因为所有3种原料药都在同一车间和生产线(103车间,C线)制造。
被EDQM采取措施的CEP
欧洲药品质量管理局(EDQM)网站更新了2019年下半年被采取措施的CEP证书。其中暂停12个CEP证书,原因分别为:“应持有人的要求暂停,因为暂时没能力在批准的条件下生产”、“GMP不符合性”,以及“未能满足认证程序的要求”,其中涉及两家中国企业。因违反GM合已被EDQM撤销CEP证书的企业有两家。详见下表:
暂停的CEP证书
应持有人的要求暂停,因为暂时没能力在批准的条件下生产
|
日期
|
品名
|
CEP编号
|
|
2019-11-5
|
水杨酸甲酯
|
CEP 2004-060
|
由于不符合GMP
|
日期
|
品名
|
CEP编号
|
|
2019-11-21
|
苯酰甲硝唑
|
CEP 2007-056
|
|
2019-11-5
|
无菌头孢噻肟钠
|
CEP 2014-197
|
|
2019-11-5
|
无菌头孢呋辛钠
|
CEP 2014-021
|
由于不满足CEP认证的条件
|
日期
|
品名
|
CEP编号
|
|
2019-8-11
|
盐酸雷尼替丁
|
CEP 2001-228
|
|
2019-10-30
|
盐酸雷尼替丁
|
CEP 2007-320
|
|
2019-10-11
|
盐酸雷尼替丁
|
CEP 1996-102
|
|
2019-10-11
|
盐酸雷尼替丁
|
CEP 2000-342
|
|
2019-10-11
|
盐酸雷尼替丁
|
CEP 2002-075
|
|
2019-10-11
|
盐酸雷尼替丁
|
CEP 2017-068
|
|
2019-9-19
|
盐酸雷尼替丁
|
CEP 2004-057
|
|
2019-7-3
|
格列吡嗪
|
CEP 2008-210
|
被撤销的CEP
由于不符合GMP
|
日期
|
物质名称
|
CEP编号
|
|
2019-11-14
|
红霉素
|
CEP 2009-267
|
|
2019-11-14
|
富马酸比索洛尔
|
CEP 2013-342
|
来源:详情链接
指南更新
EDQM更新两个CEP程序指南
CEP程序《认证方针文件和指南》集中有两个指南不再符合当前实践, 因此,EDQM做了修订。这两个指南均适用于CEP认证程序工作人员。 今年7月,发布了更新版:
-
《职权范围(PA / PH / CEP(01)1,11 R Cor)》
该文件描述了参与CEP认证程序的部门和人员要满足的要求。
原文: 详情链接
-
《认证程序操作规范(PA / PH / CEP(02)04 3R)》
该文件包含认证过程中所有工作人员的行为准则。
包含以下修订:
-
CEP审评员由CEP认证部指导委员会任命。审评员资质:
-
具有上市许可或CEP审评经验的科学家,来自监管机构,或向监管机构提供过咨询意见
-
EDQM认证部的人员,具有CEP审评经验。
-
修订版文件中不再提及来自官方药品检验实验室(OMCL)的员工。
-
关于利益冲突,对审评员和检查员实行更严格的规定。对于有利益冲突的公司,他们的活动受限制。
-
每个CEP审评和现场检查均由两名审评员和两名检查员组成的团队进行。应就结论达成共识。审评员和检查员的所有活动均需经过CEP认证部的同业互查。
总体上,这些修订内容清楚地规范和确保审评员和检查员的独立性,以避免利益冲突。
原文: 详情链接
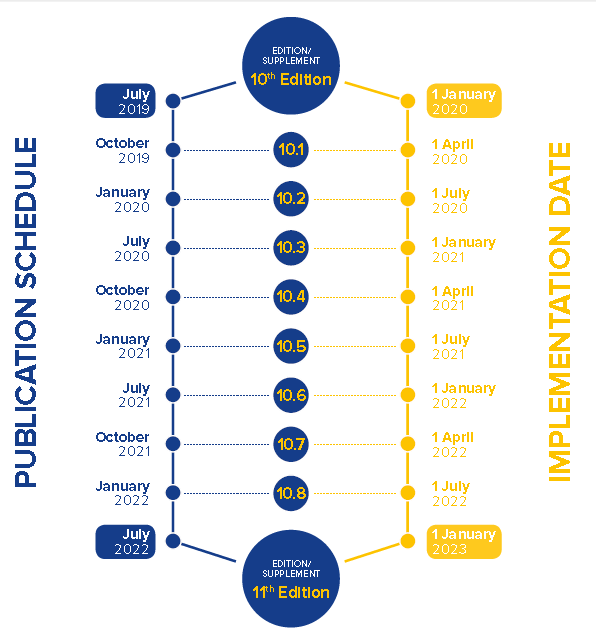
《欧洲药典》第十版于2020年1月1日执行
来源:EDQM
亚硝胺杂质检验引发《欧洲药典》第 2034篇修订稿公开征求意见
12月13日欧洲药典委员会拟修订药用物质通项(第2034篇)。该提议是基于2019年4月2日欧洲委员会第C(2019)2698号转引和欧洲药品管理局于2019年9月发起的审查依据第EC 726/2004号法规第5(3)条进一步提出的,旨在指导上市许可持有人如何避免在人用药中出现亚硝胺杂质。
因此,欧洲药典药用物质通项将进行修订,以在“生产”章节涵盖对制造工艺进行风险评估的要求,并实施控制策略用于检测和控制亚硝胺杂质。
鉴于此变更的影响,欧洲药典委员会拟将这些要求应用于所有药用物质,并征求欧洲药典用户的意见,包括以下范围:
-
活性物质(或中间体,如经论证)或辅料
-
人用或兽用物质
-
化学合成的或从天然来源中获得的,或从原料中提取的或发酵生产的
修订后的通项将发布在第32.1期在线版欧洲药典草案中,以征求公众意见,时限为2020年1月至3月。如果未收到响应,所拟修订将被认为是可接受的。
来源: 详情链接
EMA关于二甲双胍糖尿病药物的更新
12月6日,欧洲药品管理局(EMA)发布新闻稿称 EMA已知晓在欧盟境外的少数二甲双胍糖尿病药物中发现了痕量的N-亚硝基二甲胺(NDMA)杂质。
受影响的欧盟境外二甲双胍药物中NDMA含量很低,似乎处于甚至低于人们可能从其他来源(包括某些食物和水)接触的水平范围。
目前,尚无数据表明欧盟二甲双胍药物受到影响。欧盟药监机构正在与各相关企业合作,以检验欧盟的药品,在有更多信息可用时,将发布更新。
欧盟地区的患者应继续正常服用二甲双胍药物。得不到足够的糖尿病治疗的风险远远超过在检验中发现的低水平NDMA可能产生的影响。医护专业人员应提醒患者控制糖尿病的重要性。
来源:详情链接
世界卫生组织
更新的指南
新版《数据完整性指南》(征求意见稿)
10月,世卫发布了新版《数据完整性指南》(征求意见稿)。数据治理与数据完整性(DI)是确保从药品生产和控制期间所获数据和信息的可靠性的重要因素。这些数据和信息应完整,同时应可归属、清晰、同步、原始和准确,一般称为符合“ALCOA”原则。
近年,在 各类药品质量管理规范(GMP、GCP 和 GLP )检查中,数据完整性、文件记录管理实践方面的缺陷数量大大上升。可能的原因包括:(1)过于依赖人员操作,(2)使用了未进行恰当管理和验证的计算机化系统,(3)未充分审核和管理原始数据与记录。
降低此类风险需要有质量风险管理(QRM)、控制策略和科学合理原则。控制实例包括但不仅限于:
-
制订和实施 数据完整性 方针;
-
制订和实施有利于符合数据完整性要求和预期的规程;
-
在公司内采纳一种鼓励员工对失败透明化的质量文化,包括报告机制;
-
应用 质量风险管理,通过数据完整性风险评估识别所有领域的数据完整性风险,在数据整个生命周期实施适当控制,以消除或降低风险至可接受水平;
-
确保有足够的资源监测 数据完整性方针、各规程和流程的遵守情况,促进持续改进;
-
为员工提供必要的培训,例如,质量管理规范(GXP)、计算机化系统和 数据完整性;
-
根据既定用途实施和验证计算机化系统;
-
对于作为合同给出方和接受方签署的质量协议与合同,定义和管理适当的角色与职责。
详见: 详情链接
活性药用成份主文件(APIMF)申请资料变更指南修订
10月18日,世卫修订了用于支持已通过预认证的制剂或原料药的活性药用成份主文件变更指南,修订内容如下:
-
2a类变更,关于生产企业名称或地址变更,分为两个子类:1指中间体或原料药生产企业相关变更;2a.2指起始物料生产企业相关变更。
-
增加11g【AIN】类变更,用于提交元素杂质风险评估。针对原料药,与ICH Q3D指南的使用有关的文件已做相应修订。
-
关于提交原料药主文件(APIMF)新申请版本的措辞已修订,以澄清24个月期限指从APIMF接受日起计算,或从最新APIMF版本接受日起计算。
指南原文: 详情链接
世卫预认证企业名单更新
世卫更新了2019下半年通过预认证的原料药和制剂名单。其中中国企业如下:
|
参考号
|
名称
|
申请人
|
日期
|
|
原料药
|
|
WHOAPI-363
|
洛匹那韦
|
上海迪赛诺化学制药有限公司
|
2019-06-11
|
|
WHOAPI-350
|
地瑞那韦(乙酸盐)
|
浙江江北药业有限公司
|
2019-06-19
|
|
WHOAPI-261
|
吡喹酮
|
绍兴民生医药股份有限公司
|
2019-12-19
|
|
制剂
|
|
MA131
|
双氢青蒿素哌喹,薄膜包衣片剂,40mg/320mg
|
桂林南药股份有限公司
|
2019-11-19
|
|
MA139
|
双氢青蒿素哌喹,分散片 40mg/320mg
|
桂林南药股份有限公司
|
2019-11-19
|
|
MA140
|
双氢青蒿素哌喹,薄膜包衣片剂,80mg/640mg
|
桂林南药股份有限公司
|
2019-11-19
|
|
MA141
|
双氢青蒿素哌喹,分散片20mg/160mg
|
桂林南药股份有限公司
|
2019-11-19
|
|
RH089
|
米非司酮+米索前列醇,片剂,200mg + 200mcg
|
华润紫竹药业有限公司
|
2019-11-19
|
完整名单:
原料药: 详情链接
制剂:详情链接
其他组织及机构
国际标准化组织(ISO)
《纯化水和注射用水预处理和生产系统》指南
2019年6月,国际标准化组织发布了ISO 22519:2019《纯化水和注射用水预处理和生产系统》,详细说明了纯化水(PW)和注射用水(WFI)预处理和基于膜处理的生产系统的设计、材料选择、构造和运行。由于原水类型可能有多种的,因此提出了不同的组件和配置。 提供了一个决策矩阵,为不同类型的原水提供指导。一些重要章节包括:
-
3 用户需求规范(URS) 的范围
-
1 推荐的系统组件/处理阶段
-
3 构造材质一般要求
-
1 取样原则
-
2 最少采样点和位置
-
3 电导率取样
-
1 至少需要安装的仪器
-
2 在线仪器的参数监测、报警、存储和绘图
-
1 生产
-
3 消毒
-
4 余氯和氯化
-
1标准操作规程
-
2 过滤器更换
-
3 氯检验仪器
-
1 特定GMP要求概述
-
3调试和确认要求
-
1安装所需的最少控制环路
-
1 要求的报警
原文:详情链接
ISO 14644-3:2019《洁净室及相关受控环境第三部分:测试方法》
8月,ISO发布了新版ISO 14644-3:2019《洁净室及相关受控环境-第三部分:测试方法》,取代2005年版本。该文件提供了支持洁净室和洁净区运行的测试方法,以满足空气洁净度分类、其他洁净度属性和相关的受控条件。
对两种类型的洁净室和洁净区规定了性能测试:具有单向流的洁净室和具有非单向流的洁净室,均涵盖三种状态:竣工、静态和动态。
该文件提供了用于确定性能参数的测试方法、推荐的测试设备和测试程序。 如果测试方法受洁净室或洁净区类型的影响,则建议采用其他程序。
对于某些测试,建议采用几种不同的方法和设备以适应不同的最终用途。 客户和供应商之间可以通过协议使用本文中未包含的替代方法。 替代方法不一定需要提供等效的测量。
原文:详情链接
国际制药工程协会(ISPE)
ISPE 与 PDA 联合发布《提高制药场所质量文化指南》
国际制药工程协会(ISPE)与美国注射剂协会( PDA) 于10 月 7 日联合发布了《改进制药设施质量文化指南》,该指南识别了质量体系、质量文化的具体方面,并且建议了一些工具、技术和流程。
该指南旨在支持各相关组织实施一种更结构化的方式来实施调查,以提高患者安全性,并为企业创造价值。 根本原因分析(RCA)是确定问题的根本原因以识别适当的纠正或预防措施(CAPA)的过程,是任何组织持续改进计划的重要组成部分。
在CAPA系统中常见的一个问题是缺乏用于识别失败事件真正根源的严格性。这会导致这样情况,即对一起给定失败事件的“影响”或“起因”(可能阻止该事件或减轻其后果的那些因素)得到了识别和纠正,而并非根本原因的识别和纠正。这些症状通常更明显和更易于报告,而问题的实际根本原因可能被掩盖,是未知的,因此未得到解决。不足为奇地,这通常导致这类失败在其他时间或其他情况下再次发生。
指南原文:详情链接
ISPE良好实践指南:空调系统和工艺设备空气过滤器
12月国际制药工程协会(ISPE)发布了《ISPE良好实践指南:空调系统和工艺设备空气过滤器》,旨在为制药行业过滤器的选择、应用、标准、测试以及操作和维护提供参考,是《ISPE良好实践指南:空调(HVAC)系统指南》的补充。
它描述了当前技术和与当前指南相关的应用,还说明了在制造过程和安装后进行的过滤器测试的原理和目标,并包括一种用于评估生命周期成本对过滤器选择影响的建议方法。
原文:详情链接
欧洲活性药用成份委员会(APIC)
起始物料(RSM)供应商第三方审计问与答
2019年12月,欧洲化学工业协会下属的活性药用成份委员会发布了《起始物料供应商第三方审计问与答》文件。涵盖以下问题的解答:
-
本指南是否为强制性的或有法律约束力?
-
我们是否需要审计注册的起始物料供应商?
-
是否只能由质量部门人员执行审计?
-
如果一个供应商拒绝现场审计,而根据你们的规程和/或风险评估必须进行审计,将如何处理?
-
是否需要审计分销商以代替供应商审计?
-
如果供应商不提供制造工艺的信息,将如何处理?
-
如果起始物料供应商没有质量部门,将如何处理?
-
对合同人的培训是否需要文件化?
-
是否允许在“开放式”或“部分开放式”环境中进行生产操作?
-
如果政府部门(没有原始数据)发了校准证书,是否可认为设备已校准?
-
起始物料供应商是否必须对所有进厂原料进行检验?
-
是否需要执行ICH 稳定性试验以确定有效期或复验期?
-
起始物料供应商是否需要对起始物料检验的分析方法进行验证?
-
起始物料是否可在待验状态发运?
原文:详情链接
注:RSM全称“registered starting material”,注册的起始物料。
国际药品认证合作组织(PIC/S)
PIC/S 2020年工作计划
国际药品认证合作组织于11月11-12日在日本富山举行了会议,随后发布了2020年工作计划。
PIC/S将与欧洲药品管理局(EMA)的检查员工作组紧密合作,以进一步修订PIC/S GMP指南。通常,PIC/S根据EMA-PIC/S联合磋商程序通过检查员工作组的专家参与该工作。2020年,将继续以下修订工作:
|
GMP指南
|
主题
|
|
第一章
|
制药质量体系
|
|
第四章、附录十一
|
文件、计算机化系统
|
|
附录一
|
无菌医品
|
|
附录二
|
人用生物药活性成分&成品
|
|
附录四、五*
|
兽用医疗产品和生物制品
|
将进一步修订或开发以下相关指南:
|
编号
|
主题
|
|
PE 005-3
PI 008-3
PI 019
PI 020
|
修订PIC/S 血液设施GMP指南(PE 005-3)
PIC/S血浆设施和血浆仓库检查指南(PI 008-3)
PIC/S源血浆设施场地主文件(PI 019)
PIC/S血浆仓库场地主文件(PI 020)
|
|
PE 010-4
|
PIC/S医疗场所医用产品GMP(在注射用营养剂(TPN)总指南中增加附录)
|
|
PI 006-3
|
修订PIC/S验证主计划、安装和运行确认、非无菌工艺验证和清洁验证
|
|
PI 007
|
无菌工艺验证建议
|
|
PI 011
|
PIC/S受监管的GXP环境下计算机化系统GMP
|
|
PI 013
|
PIC/S检查报告
|
|
PI 023-2
|
QC实验室检查备忘
|
|
PI 030-1
|
API检查备忘
|
|
PI 049
PI 050
|
PIC/SGMP/GDP环境下数据管理和完整性优良规范指南
数据管理和数据完整性备忘
PIC/S数据完整性系统特定指南备忘:Chromeleon 7色谱数据系统和服务器/客户系统
|
|
PI 052
PI 053
|
共用设备交叉污染备忘
共用设备交叉污染检查备忘
HBEL暴露限和HBEL问与答备忘
|
原文:详情链接
|


